Key Message
-
This retrospective study reviewed 22 children diagnosed with hemophagocytic lymphohistiocytosis (HLH).
-
It characterizes the clinical features, genetic profiles, laboratory findings, and neurodiagnostic studies in HLH patients with neurological involvement.
-
Central nervous system (CNS) involvement in pediatric HLH is associated with significant morbidity and mortality.
-
Implementing institutional protocols to screen children with HLH for CNS involvement may improve survival outcomes.
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening hyperimmune condition triggered by the pathological activation of T cells, natural killer cells, and macrophages, leading to a cytokine storm and widespread hyperinflammation. This condition often affects multiple organs, including the liver, lymph nodes, spleen, and brain.1
Historically considered rare, HLH has an estimated incidence of 1.2 cases per million children, with 40% of cases occurring in adults.2–4 Approximately 1 in 3000 patients admitted to pediatric tertiary centers may have HLH.5 Current diagnostic and management guidelines are based on the HLH 2004 study conducted by the Histiocyte Society.6 Patients must meet five out of eight diagnostic criteria established by the HLH-2004 group (Table 1).7 While not part of the formal diagnostic criteria, many HLH patients exhibit central nervous system (CNS) signs and symptoms.
Familial HLH (fHLH), also known as primary HLH (pHLH), is caused by homozygous or compound heterozygous mutations in genes involved in the cytotoxic function of T cells.1,8 Mutations in PRF1, UNC13D, STX11, STXBP2, Rab27, SH2D1A, BIRC4, LYST, ITK, SLCA7, XMEN, and HPS are common causes of pHLH, with inheritance patterns being autosomal recessive or X-linked.1,9,10 Secondary HLH (sHLH) can be triggered by infections, malignancies, or underlying rheumatologic conditions, with Epstein-Barr virus (EBV) infection often associated with severe cases.11–14
CNS involvement in HLH is characterized by the aberrant activation and infiltration of inflammatory cells into the leptomeninges, brain parenchyma, and perivascular spaces, leading to a cytokine storm that can cause demyelination, parenchymal necrosis, and calcifications.8,14–20 Neurologic manifestations vary and may include generalized weakness, altered sensorium, headaches, seizures, cranial nerve palsies, ataxia, irritability, altered consciousness, and hypotonia.21–25 Neuroimaging changes may precede neurologic symptoms, though the timeline of CNS involvement remains unclear.26 CNS involvement occurs in 30-70% of HLH cases and can be the sole clinical presentation.21–25 Cerebrospinal fluid (CSF) pleocytosis and modest protein elevation are observed in 10-47% of patients.27–30 Neuroimaging findings include nonspecific brain atrophy, multifocal symmetric periventricular white matter abnormalities, calcifications, focal cortical/subcortical lesions, and hemorrhage.14,18,19
HLH can be fatal without treatment. Current therapy for pHLH includes etoposide and dexamethasone, with intrathecal methotrexate added for CNS involvement.5,31 Patients with identified pHLH mutations require hematopoietic stem cell transplantation (HSCT), which, if performed early, may halt CNS disease progression and prevent relapses.5,32 Many pediatric and adult patients with sHLH lack identifiable genetic mutations and are not candidates for HSCT.
Despite established diagnostic criteria, diagnosing HLH with CNS manifestations (CNS-HLH) remains challenging, as it can mimic conditions like acute disseminated encephalomyelitis, septic shock, malignancy, and hyperinflammatory syndromes such as multisystem inflammatory syndrome in children (MIS-C).33–35 Although outcomes have improved with allogeneic bone marrow transplantation,36 CNS-HLH continues to carry a high burden of morbidity and mortality, with motor and cognitive deficits and a 5-year survival rate of 40-67%.16,21 The true burden of CNS disease in sHLH and its optimal management remain unclear.
Primary aim
This study aimed to characterize CNS features in pediatric HLH patients.
Methods
This retrospective chart review included pediatric patients hospitalized with HLH from August 26, 2013, to September 5, 2020, at a single medical center. Patients were followed for up to 12 years. Data were collected from electronic medical records after Institutional Review Board approval.
Patients were identified using the ICD-10 code for HLH. Of 48 children, 26 were excluded for not meeting HLH-2004 diagnostic criteria or lacking a molecular diagnosis. Patients with prior HLH diagnoses not primarily hospitalized at the institution were also excluded. Inclusion criteria were pediatric patients meeting HLH-2004 diagnostic criteria, including both pHLH and sHLH cases.
Data collected included demographics, genetic test results, physician reported history of symptoms and clinical signs, CSF and blood viral panels and bacterial cultures.
Laboratory results included initial, maximum, and minimum values of hemoglobin, platelets, absolute neutrophil count, ferritin, C-reactive protein, erythrocyte sedimentation rate, aspartate transaminase, alanine transaminase, gamma-glutamyl transpeptidase, triglycerides, fibrinogen, lactate dehydrogenase, direct and indirect bilirubin, and interleukin-2 receptor antibody levels.
CNS involvement was defined by neurologic symptoms and signs, abnormal CSF studies, neuroimaging, or EEG. CSF abnormalities included leukocyte counts >5 cells/mL or protein >50 mg/dL. Brain MRI findings, reviewed by a neuroradiologist, included diffuse atrophy, T1/T2 abnormalities, diffusion restriction, micro-/large hemorrhages, or calcifications. EEG reports were reviewed for focal or generalized slowing and epileptiform abnormalities.
Statistical analysis
Data were entered into REDCap and analyzed using IBM SPSS Statistics for MacBook (version 28.0.1.0). Categorical variables were compared using Chi-square or Fisher’s exact tests, and non-parametric continuous variables were analyzed using the Mann-Whitney U test, reported as medians with interquartile ranges. A p-value <0.05 was considered significant.
Results
Twenty-two children were included. Please refer to Table 2 for detailed demographic information. Genetic testing was performed in 20 patients, with 6 (30%) having pathogenic mutations in pHLH genes, classifying them as pHLH. The remaining 14 were classified as sHLH, with 8 having variants of unknown significance and 6 having no variants. The clinical characteristics and CNS findings of these pHLH patients are summarized in Table 3.
Four (18.2%) children had coexisting malignancies. No significant differences were observed in infection, sepsis, meningitis, multi-organ dysfunction, stroke, or hematologic/rheumatologic abnormalities between survivors and deceased patients. Details regarding coexisting conditions can be seen in Table 5.
17 (77.2%) were noted to have CNS involvement. Neurology consultations were obtained for 8 (36.3%) patients at a median of 26 days from symptom onset.
Seventeen (77.2%) patients developed CNS symptoms during hospitalization. Symptoms were overlapping and included generalized weakness 11 (30.3%), altered sensorium 8 (47%), seizure/seizure-like activity 4 (23.5%), headaches 3 (17.6%) and focal weakness with vision abnormalities 1 (5.8%).
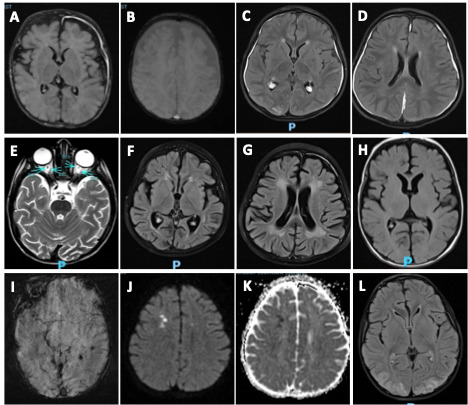
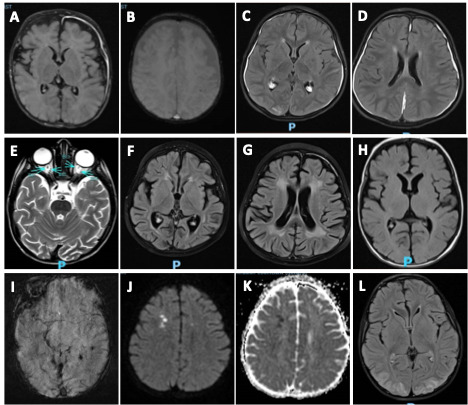
MRI scans were obtained in 20 of 22 (90.9%) patients at a median of 17 days from symptom onset with overlapping findings. Patchy T2 FLAIR prolongation was noted in 17 (85%) patients with 6 (35.2%) patients having T2 FLAIR prolongation in gray matter. Other MRI abnormalities included diffuse brain atrophy 8 (40%), microhemorrhage 3 (15%) and diffusion restriction 3 (15%). MRI abnormalities in pediatric pHLH patients are shown in Figure 1.
6 (27.2%) patients received an EEG with 5 (83.3%) patients showing mild/moderate generalized polymorphic slowing suggesting etiologically non-specific diffuse cortical dysfunction, 1 patient exhibiting interictal epileptiform discharges and 1 patient with a recorded electrographic seizure.
No significant differences were observed in laboratory values between patients with and without CNS involvement. Please refer to supplemental Table 6 for details. CSF studies in 17 patients revealed a median WBC count of 15.5 cells/mL in pHLH compared to 1 cell/mL in sHLH (p=0.005). Median CSF values with interquartile ranges are detailed in Table 7.
Of 17 patients with CNS involvement, 8 (47.05%) received dexamethasone, etoposide, and cyclosporine; 6 (35.2%) received dexamethasone alone; 2 (11.7%) received intrathecal methotrexate; and 5 (29.4%) underwent HSCT. Other treatments included IV immunoglobulins, anakinra, anti-thymocyte globulin, and rituximab.
In the initial one year, 4 (18%) children died, with 3 (75%) having CNS involvement. Mean survival duration at six months was 167 days. No further deaths occurred during the 12-year follow-up. See Table 4 for details regarding deceased HLH patients with CNS involvement).
Discussion
This study describes CNS features in 22 pediatric HLH patients over seven years. CNS involvement was determined by neurologic symptoms and abnormal neurodiagnostic studies, with 77% of patients demonstrating CNS involvement. Generalized weakness and altered sensorium were the most common neurologic symptoms with 23% patients noted to have seizures. This is consistent with previous studies.21–25,30,37
CNS symptoms likely result from meningeal and parenchymal inflammation due to lymphocytic and macrophage infiltration.38 One patient with pHLH exhibited an acute stroke that resolved but later developed posterior reversible encephalopathy syndrome (PRES) due to HLH reactivation.
CSF findings of increased WBC counts are consistent with findings of Shyu et al.39 The basis of CSF pleocytosis is likely related to the aberrant neuroinflammatory response involving a dysregulated cytokine and interleukin cascade.
T2 FLAIR prolongation in gray matter was frequently observed, possibly due to impaired mitochondrial oxidative phosphorylation or global hypoperfusion.40 Brain atrophy, microhemorrhages, white matter abnormalities, and leptomeningeal enhancement were also noted, consistent with prior findings.14,41 Higher CSF WBC count in patients with pHLH compared to sHLH may be due to difference in pathophysiology. The underlying genetic mutation in pHLH can produce a more pronounced aberrant activation and infiltration of macrophages and microglia in the CNS, resulting in higher degree of CSF pleocytosis.
Treatment followed HLH 2004 protocols, with dexamethasone, etoposide, cyclosporine, and intrathecal methotrexate used for CNS involvement. HSCT was pursued early. Variability in management reflects the study’s retrospective nature and evolving practices over time.
Mortality was 18%, primarily occurring within the first nine months. Of the two patients who received HSCT and then died, one patient developed pneumonia (post HSCT) and subsequent respiratory failure leading to demise, and the other developed CMV viremia and ARDS leading to demise.
Limitations
The retrospective design and management variability over seven years may have influenced results. Defining CNS involvement based on symptoms and neurodiagnostic studies may not capture all cases. The rarity of HLH limits generalizability, and larger studies are needed to differentiate pHLH and sHLH.
Conclusion
This study highlights the significant CNS involvement (77%) in pediatric HLH patients. Early and aggressive management may alter the disease course. Newer therapies such as ruxolitinib may yet also alter the natural history of this disease. Treating clinicians should remain vigilant in surveilling for, recognizing and treating CNS involvement in HLH patients.
Author Contributions
Conceptualization: Manan Nath (Equal), Anshul Vagrecha (Equal), Sanjeev V. Kothare (Equal). Methodology: Manan Nath (Equal), Anshul Vagrecha (Equal), Sanjeev V. Kothare (Equal). Formal Analysis: Manan Nath (Equal), Anshul Vagrecha (Equal), Sanjeev V. Kothare (Equal). Investigation: Manan Nath (Equal), Anshul Vagrecha (Equal), Yash D. Shah (Equal), Robin Varughese (Equal), Ramya Trietel (Equal), Alan Johnson (Equal), Carolyn Fein-Levy (Equal), Sanjeev V. Kothare (Equal). Writing – original draft: Manan Nath (Equal), Anshul Vagrecha (Equal), Carolyn Fein-Levy (Equal), Sanjeev V. Kothare (Equal). Writing – review & editing: Manan Nath (Equal), Annie H. Roliz (Equal), Carolyn Fein-Levy (Equal), Sanjeev V. Kothare (Equal). Resources: Manan Nath (Equal), Anshul Vagrecha (Equal), Carolyn Fein-Levy (Equal), Sanjeev V. Kothare (Equal). Supervision: Sanjeev V. Kothare (Lead).
Support/grant information
No funding was granted for this manuscript
Conflict of Interest Statement
The author(s) declare(s) that there is no conflict of interest.
Financial Disclosures
Dr. Nath, Dr. Roliz, Dr. Shah, Dr. Treitel, Dr. Varughese, Dr. Johnson, Dr. Fein-Levy and Dr. Kothare report no disclosures