1. Introduction
VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) syndrome is caused by a somatic UBA1 mutation and often associated with myelodysplastic syndrome (MDS).1–4 It is a complex systemic disorder involving multiple hematopoietic stem cell clones, which may affect disease progression and treatment response.5 The efficacy of conventional immunosuppressive and anti-inflammatory therapies is limited, and allogeneic hematopoietic stem cell transplantation (allo-HSCT) is considered the only curative option.6,7 However, evaluation for co-mutations, optimal timing and donor source of HSCT, and post-transplant management remain unclear. To contextualize these unresolved questions, we first present a representative case. This case illustrates the challenges of refractoriness to medical treatment, complexity of clonality, and the difficulty of donor selection, which form the framework for our systematic review.
2. Case Presentation
A 67-year-old man visited his primary care physician complaining of recurrent pharyngalgia, conjunctival injection, arthralgia, and fever. Based on the finding of macrocytic anemia with a hemoglobin level of 8.0 g/dL, a hematological disorder was suspected, and the patient was referred to our department for specialized treatment.
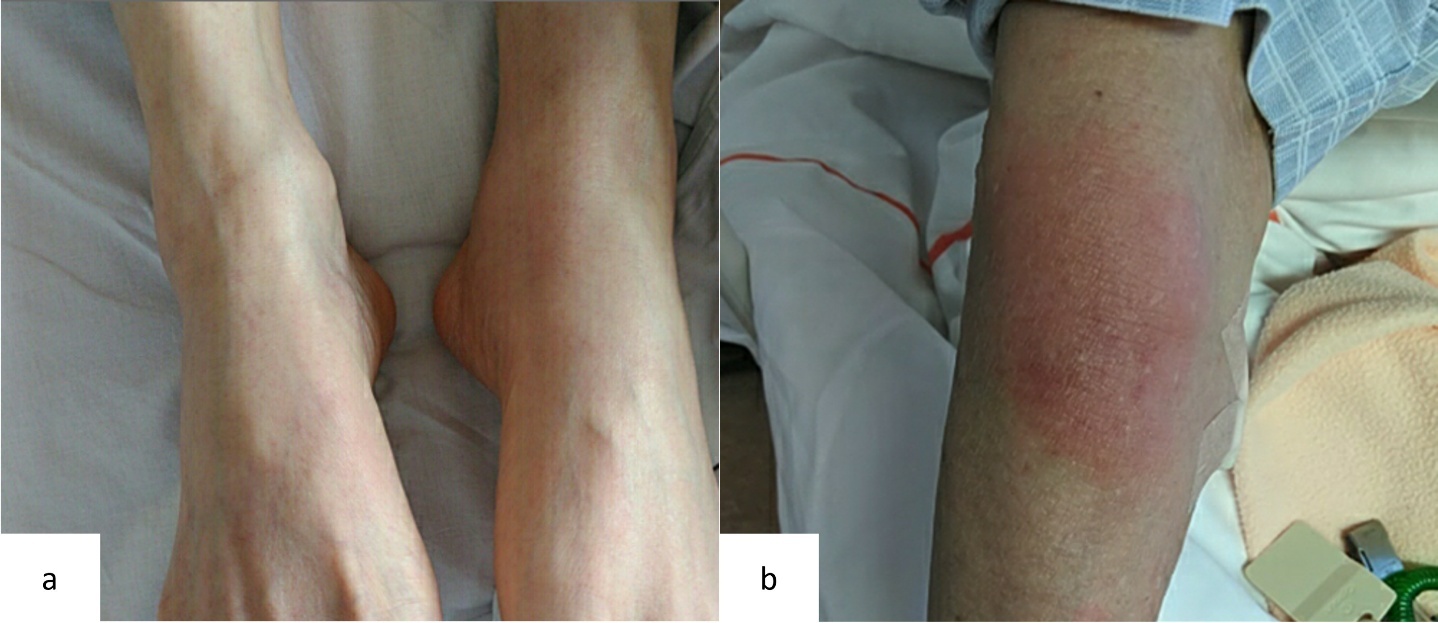
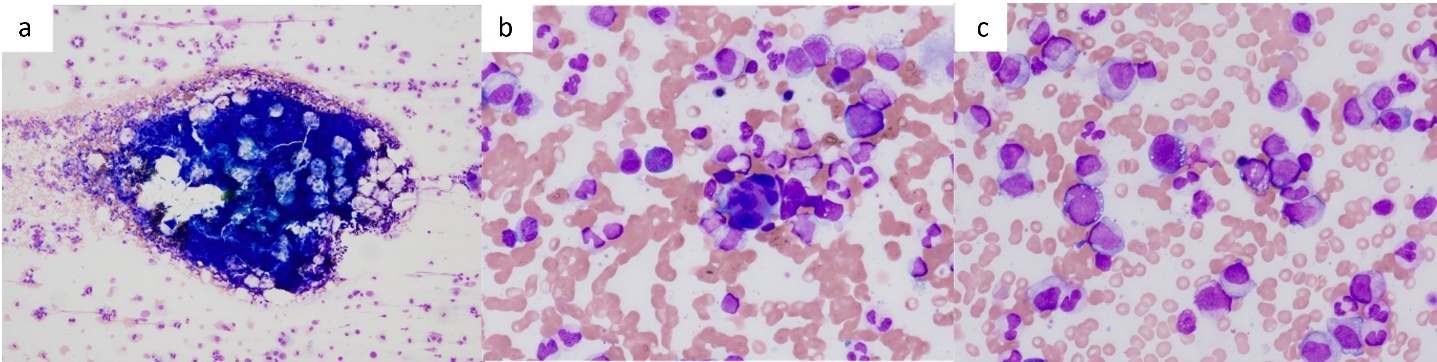
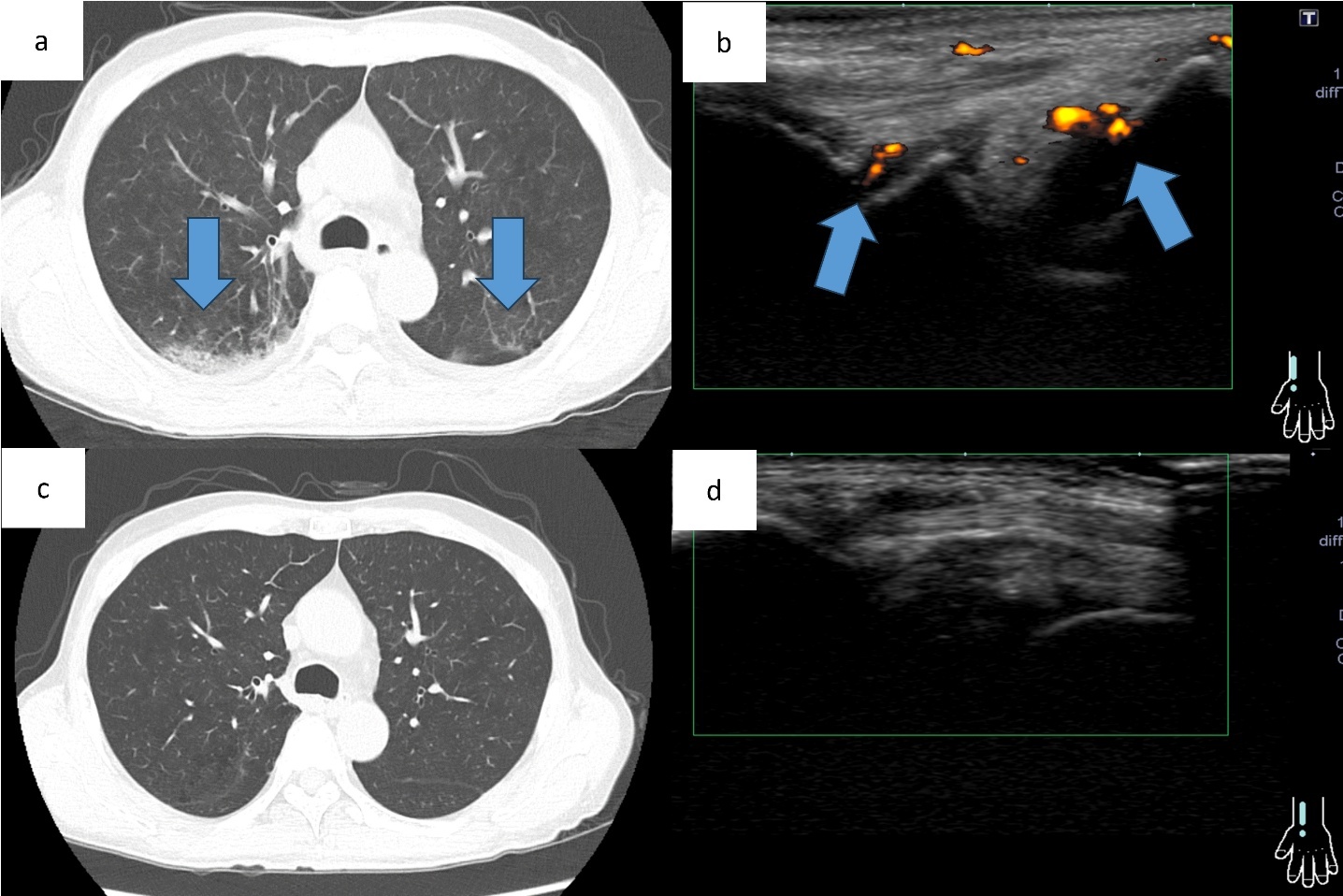
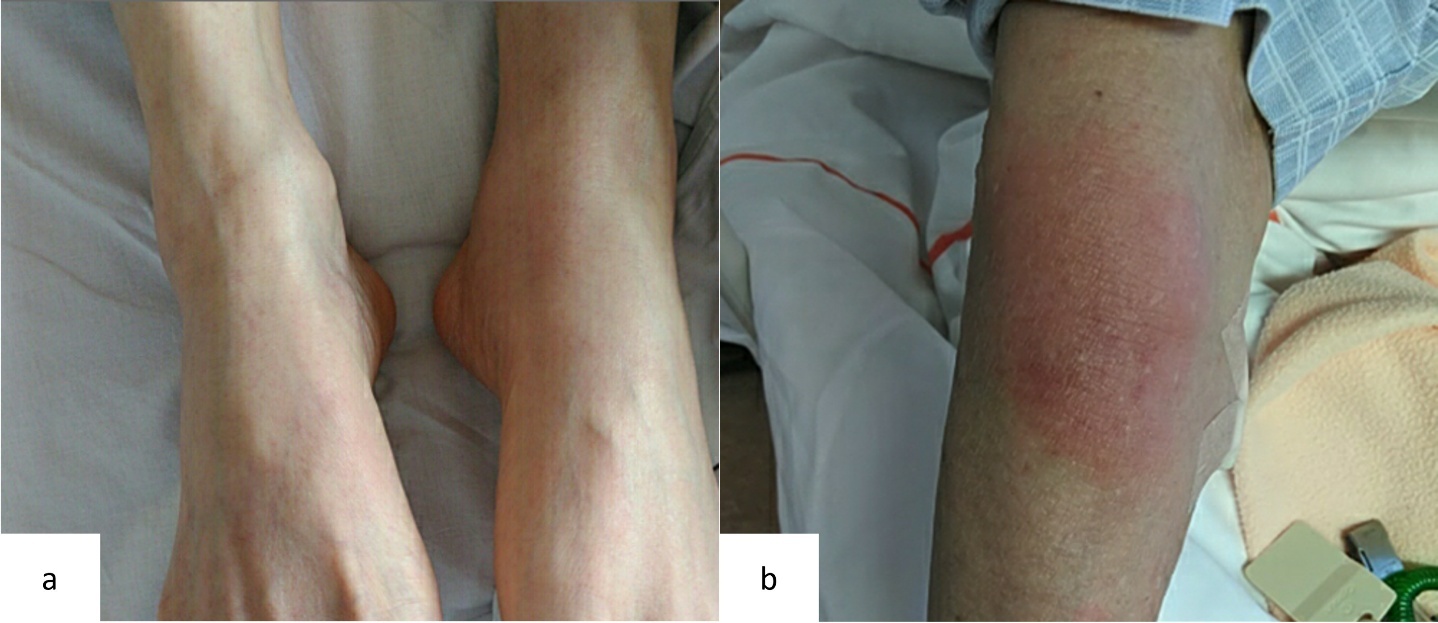
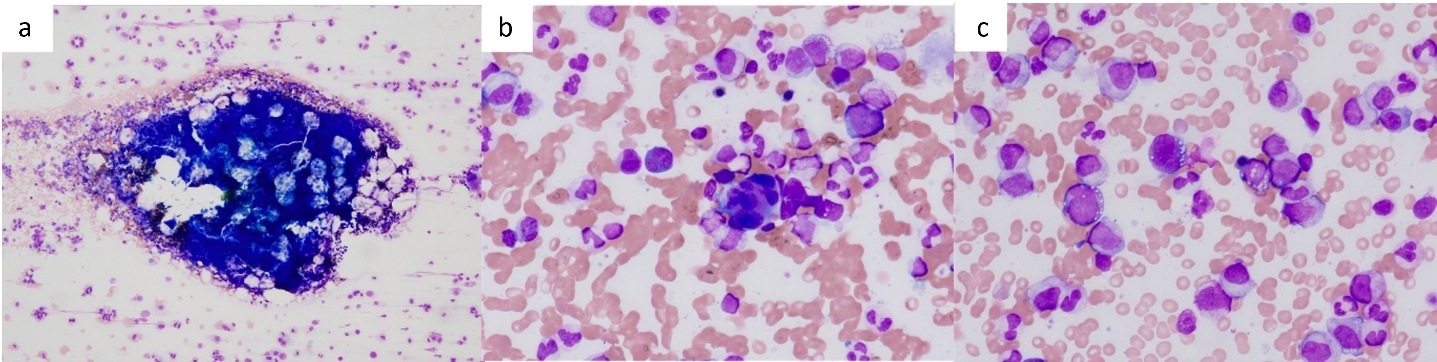
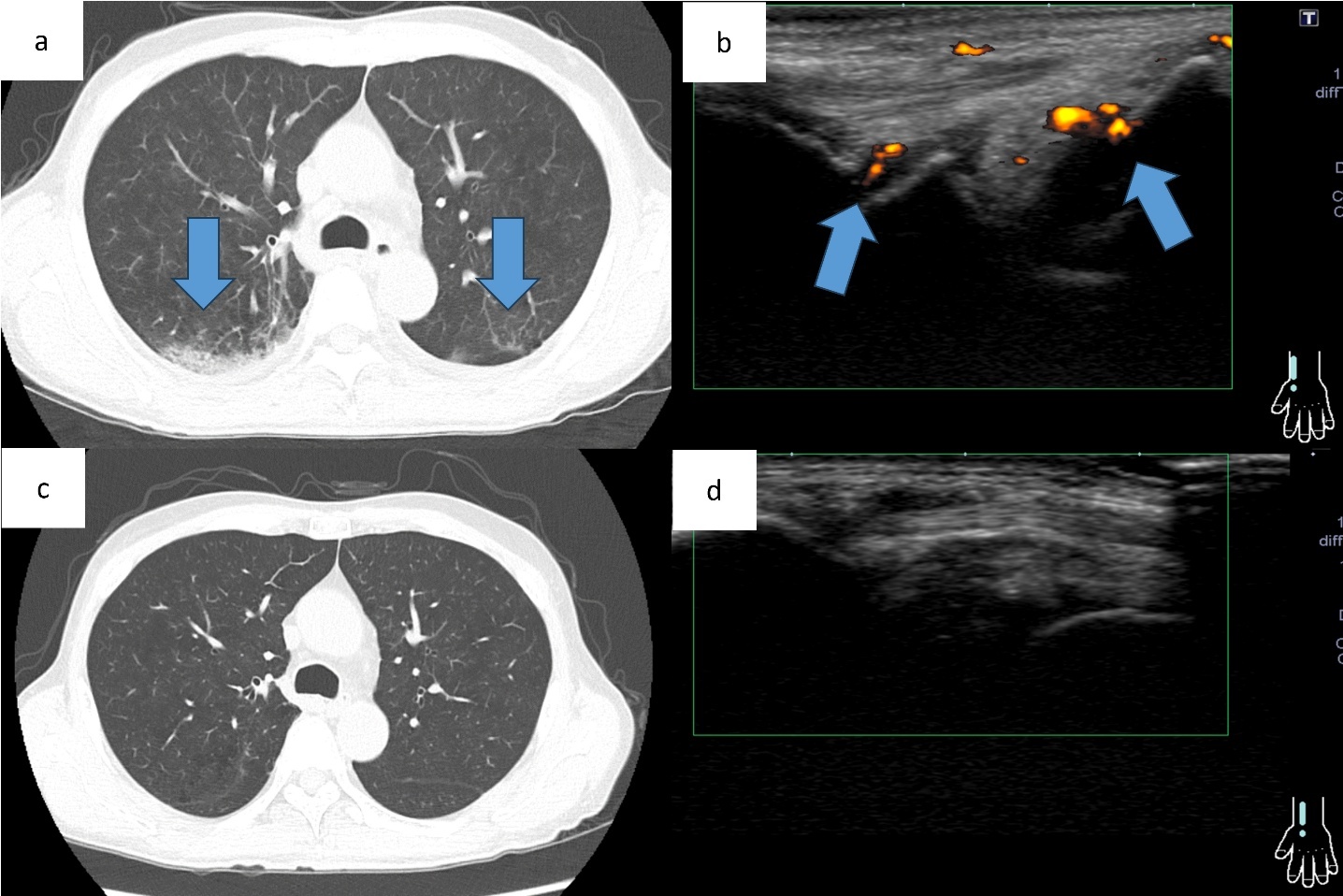
At the initial visit to our department, he complained of fever and pain in both knees. Erythema was observed on both ankles (Figure 1a), along with redness of the left auricle. Blood tests revealed elevated inflammatory markers, including an increased C-reactive protein level and macrocytic anemia. All tested autoantibodies, such as antinuclear antibodies, antineutrophil cytoplasmic antibodies, and rheumatoid factors, were negative (Table 1). Bone marrow aspiration showed hypercellularity with a nucleated cell count of 251,000/μL, trilineage dysplasia predominantly in the megakaryocytic lineage, and numerous hematopoietic progenitor cells with cytoplasmic vacuoles (Figure 2). Chest computed tomography demonstrated bilateral peripheral ground-glass opacities (Figure 3a). Joint ultrasonography revealed synovial thickening and increased blood flow in the joints (Figure 3b).
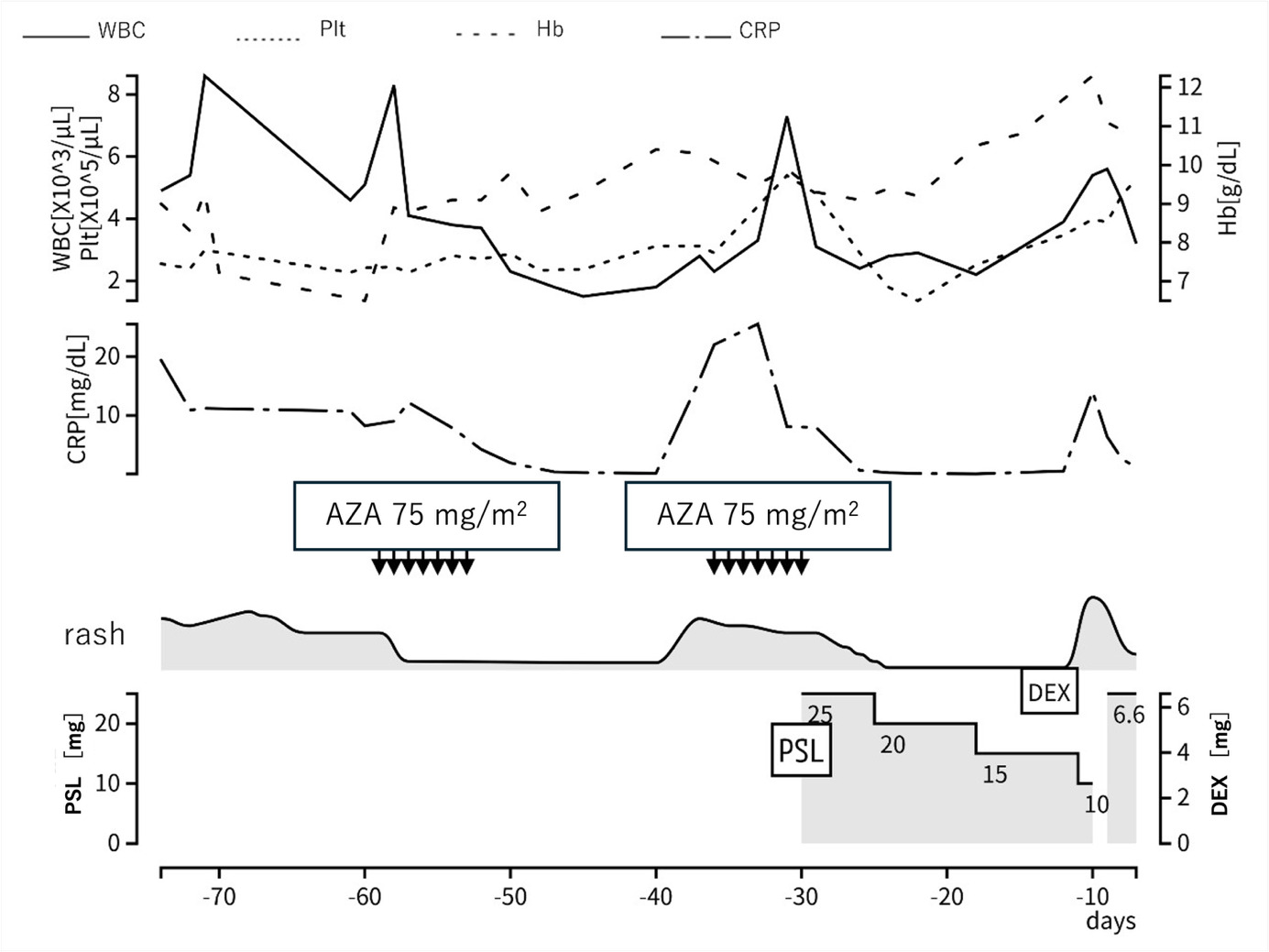
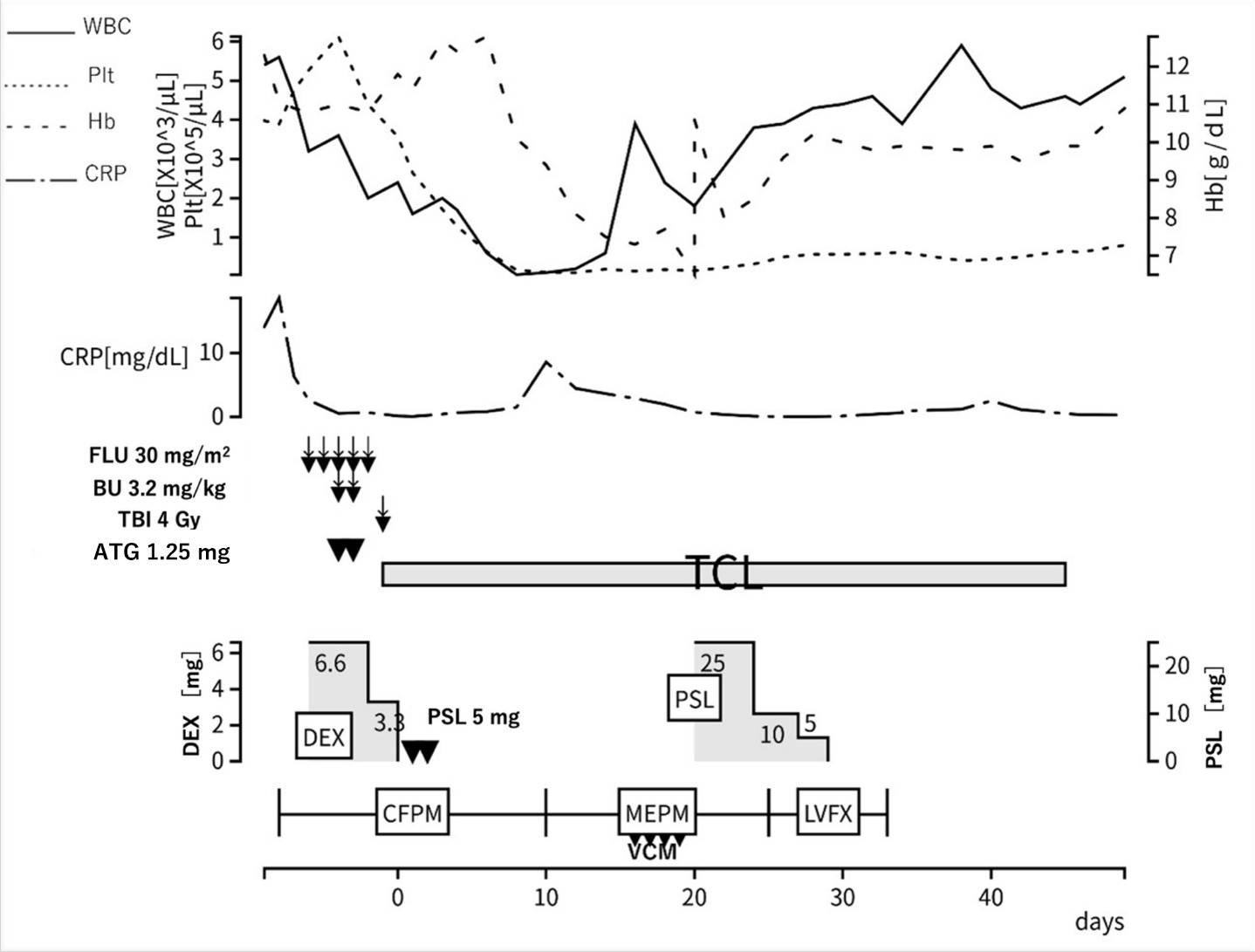
Based on these findings, a provisional diagnosis of MDS complicated by an autoimmune disorder was made. Azacitidine was started to control the disease. The initial administration of azacitidine temporarily improved the erythema and inflammatory findings (Figure 4). However, symptoms recurred, so the treatment interval was shortened to 5 days for the second course. The second course was less effective than the first course. Then, prednisolone 0.5 mg/kg was started. His symptoms improved, and the dose of prednisolone was gradually tapered. However, symptoms recurred when the dose was tapered to 10 mg/day (Figure 1b). Therefore, dexamethasone 6.6 mg was added.
.png)
At this point, comprehensive gene analysis using next generation sequencing8 performed on the bone marrow sample identified somatic mutations in the UBA1 p.M41T (variant allele frequency 0.64) and EZH2 p.N676fs (variant allele frequency 0.16) genes. Based on these findings, a diagnosis of VEXAS syndrome associated with MDS was made. Considering the resistance to azacitidine and prednisolone, allo-HSCT was planned as a curative treatment (Figure 5). The conditioning regimen consisted of fludarabine (150 mg/m²), busulfan (6.4 mg/kg), and total body irradiation (4 Gy). The patient underwent an allo-HSCT from his one human leukocyte antigen (HLA)-mismatched daughter. Tacrolimus, short-term methotrexate, and antithymocyte globulin (ATG) (1.25 mg/kg for 2 days) were administered for graft-versus host disease (GVHD) prophylaxis. The Hematopoietic Cell Transplantation-Comorbidity Index score was 4 before transplantation due to decreased respiratory and hepatic function.
.png)
Engraftment of hematopoietic stem cells was confirmed on Day 14. Engraftment syndrome developed on Day 18, but rapidly improved with the administration of prednisolone 0.5 mg/kg (25 mg). The patient’s general condition was stable, and he was discharged on Day 50. On Day 81, however, erythema appeared on his face and scalp and spread to his limbs and trunk. He was diagnosed with acute skin GVHD (stage 3), and prednisolone 0.5 mg/kg (25 mg) was restarted on Day 95, resulting in rapid improvement of the erythema. When prednisolone was tapered to 7 mg, lichen planus-like skin lesions appeared, and he was diagnosed with chronic GVHD (NIH skin score 2). Prednisolone was increased again to 15 mg on Day 172, but the skin lesions showed poor improvement. Liver dysfunction (AST 146 U/L, ALT 202 U/L, γ-GTP 1082 U/L, NIH score 1) also developed. Ruxolitinib 20 mg was introduced as the second-line treatment on Day 180, resulting in improvement of both the skin lesions and liver dysfunction. Subsequently, prednisolone was tapered to 6 mg, and there has been no GVHD recurrence.
At post-transplant follow-up, the peripheral ground-glass opacities on chest computed tomography and arthritis on joint ultrasonography observed before transplantation had resolved (Figure 3cd). Hematological counts and inflammatory markers have remained stable. To date, there have been no clinical or radiological findings suggestive of a recurrence of VEXAS syndrome.
3. Methods of systematic review
A literature search was conducted on PubMed from its inception until August 2025. The keywords “VEXAS” and “allogeneic hematopoietic stem cell transplantation” were used. Eligible studies included retrospective and prospective studies, case reports, and case series of VEXAS syndrome treated with allo-HSCT. Review articles, studies lacking data on treatment, treatment response, and individual patient data were excluded. Eight studies were selected for review, from which data on the clinical characteristics, number of pre-transplant treatment regimens, and transplant information of 45 patients were extracted (Table 2).9–17
4. Results of systematic review
4-1. Patient Cohort and Disease Characteristics
A total of 45 cases of VEXAS syndrome treated with allo-HSCT from eight studies (Table 3) were analyzed. The median age at transplantation was 59 years (range: 46–70 years). MDS was the most common underlying hematologic disorder, present in 24 cases (53.3%); 19 cases (42.2%) were treated for VEXAS syndrome alone, in the absence of overt MDS/myeloid neoplasms.
The p.Met41Val variant was the most common variant, occurring in 17 cases (37.8%), followed by the p.Met41Thr variant in 14 cases (31.1%) and the p.Met41Leu variant in 4 cases (8.9%).
4-2. Transplant Characteristics and Pre-transplant Treatment
A median of five (range: 0–13) systemic drug regimens consisting primarily of immunosuppressants were administered before transplantation. Reduced-intensity conditioning was the predominant approach in most cases. For graft-versus-host disease (GVHD) prophylaxis, post-transplant cyclophosphamide was administered in 20 cases (44.4%). Most transplants were from HLA-matched donors, but three were from haploidentical donors. Most patients received peripheral blood stem cell transplant, but two received bone marrow transplant.
4-3. Post-transplant Outcomes
At the time of reporting, 38 (84.4%) of the 45 cases were alive, in most of whom surviving clinical symptoms resolved. Ruxolitinib and belumosudil were administered for post-transplant complications, including acute and chronic GVHD, and recurrent inflammation.
5. Integrated Discussion and Treatment Strategy
5-1. Limitations of Drug Therapy and Need for Transplantation
Patients with VEXAS syndrome typically receive multiple immunosuppressants before definitive diagnosis is established.1 Although these treatments may alleviate symptoms and temporarily improve quality of life, they do not eradicate the underlying UBA1-mutant hematopoietic clone, inherently limiting their ability to achieve sustained disease control. Consistent with this limitation, our review showed that many patients had received multiple systemic therapies prior to transplantation, highlighting the refractory nature of VEXAS syndrome to medical treatment.
In the present case, azacitidine and corticosteroids resulted in only transient clinical improvement, followed by early steroid dependence. These findings suggest that long-term disease control with drug therapy alone would be difficult. Supporting this perspective, a recent meta-analysis has shown that allo-HSCT represents a reliable therapeutic option for selected patients with VEXAS syndrome.18
5-2. Transition from Drug Therapy to Transplantation
Determining the optimal timing for transition from drug therapy to allo-HSCT is one of the most challenging aspects of managing VEXAS syndrome. Initial treatment strategies typically focus on disease control using agents such as corticosteroids and azacitidine,4,5,19 but long-term disease control is often elusive, and many patients eventually develop treatment refractoriness or steroid dependence.4 Once resistance to drug therapy or steroid dependence becomes evident, allo-HSCT is regarded as the only curative option. At this stage, the therapeutic goal shifts from achieving prolonged disease control to preserving performance status and minimizing treatment-related toxicity in preparation for allo-HSCT.6
In the present case, early recognition of azacitidine resistance and steroid dependence prompted a timely transition to a transplant-oriented strategy. Consequently, allo-HSCT was performed while the patient maintained good performance status, despite his advanced age.
5-3. Importance of Personalized Treatment Based on Genetic Mutation Profiles
Emerging evidence suggests that the clinical phenotype and prognosis of VEXAS syndrome vary according to the type of UBA1 mutation.3 In our review, the p.Met41Val variant, which was associated with poor prognosis, was the most frequently observed mutation among transplanted cases. In addition to UBA1 mutations, VEXAS syndrome is frequently accompanied by clonal hematopoiesis involving additional CHIP-associated gene mutations. Gutierrez-Rodrigues et al. reported a high prevalence of mutations in genes such as DNMT3A, TET2, and ASXL1, underscoring the genetic complexity of this disorder.20
In the present case, an EZH2 mutation was identified in addition to the UBA1 mutation. This co-mutation provides a plausible molecular explanation for the observed resistance to azacitidine.21,22 These findings emphasize the importance of viewing VEXAS syndrome not as a disease driven solely by a single UBA1 mutation, but rather as a disorder arising from complex, polyclonal hematopoiesis. This understanding is crucial when formulating individualized treatment strategies, particularly with respect to assessing the limitations of drug therapy and determining the timing of allo-HSCT.
5-4. Donor Source
In allo-HSCT, an HLA-matched donor is generally considered optimal. In our review, most patients underwent transplantation from HLA-matched donors. However, because VEXAS syndrome predominantly affects the elderly, the availability of HLA-matched sibling donors is often limited. In the present case, allo-HSCT from his one-antigen HLA-mismatched daughter resulted in successful engraftment and durable remission. This suggests that transplantation may remain a feasible option even in patients with limited donor availability.
5-5. Conditioning Regimen
Patients with VEXAS syndrome are typically of advanced age and frequently have comorbidities. As a result, reduced-intensity conditioning (RIC) regimens are commonly favored. Accordingly, most cases included in our review received RIC regimens. In the present case, a RIC regimen consisting of fludarabine, low dose busulfan, and reduced total body irradiation was selected based on his age and comorbidities. At present, an optimal conditioning regimen specific to VEXAS syndrome has not been established, and further accumulation of clinical experience is required.
5-6. GVHD Prophylaxis
The primary objective of allo-HSCT for VEXAS syndrome is to replace the UBA1-mutant clone with normal donor-derived stem cells. Unlike acute myeloid leukemia, a strong graft-versus-tumor effect is not necessarily required; therefore, stable engraftment with minimal toxicity represents the highest priority.
At the time this case was treated, post-transplant cyclophosphamide (PTCy) was approved in Japan only for HLA-haploidentical transplantation, and ATG was therefore used for GVHD prophylaxis. In recent years, however, PTCy has been widely adopted for GVHD prophylaxis across various transplant settings, including one-allele HLA-mismatched and HLA-matched transplantation.23,24 Emerging data suggest that PTCy may also be useful in selected patients with VEXAS syndrome.
5-7. Post-transplant Treatment
GVHD remains a major post-transplant complication in patients with VEXAS syndrome. A recent meta-analysis reported an incidence of grade II–IV acute GVHD of 42% and chronic GVHD of 13%, underscoring the importance of effective post-transplant management.17 Ruxolitinib, a Janus kinase (JAK) inhibitor, has emerged as a promising therapeutic option in this setting.25,26 It is not only effective for steroid-refractory GVHD, but also for the inflammatory state of VEXAS syndrome.27,28 In the present case, use of ruxolitinib resulted in improvement of cutaneous and hepatic GVHD, without recurrence of VEXAS-related inflammatory symptoms. Although evidence remains limited, post-transplant ruxolitinib may contribute to improved outcomes.
5-8. What the Present Case Adds Beyond the Review
In our review of 45 patients with VEXAS syndrome who underwent allo-HSCT, the median age at transplantation was 59 years, and patients had received a median of five systemic treatment regimens prior to transplantation. Most transplants were performed using HLA-matched donors, and PTCy was used for GVHD prophylaxis in some patients.
In contrast, the present patient underwent transplantation at 67 years of age (older than the cohort median) after only two lines of pre-transplant therapy, while maintaining good performance status. A one-antigen mismatched related donor was selected, and favorable outcomes were achieved using ATG-based GVHD prophylaxis and post-transplant ruxolitinib.
This contrast suggests that early, genetics-informed decision-making based on treatment response may improve transplant outcomes even in older patients. Notably, the presence of an EZH2 mutation in this case provided a molecular rationale for resistance to drug therapy and supported the decision to proceed to allo-HSCT. This case therefore adds meaningful clinical insight to our review by illustrating individualized decision-making processes that are not readily captured by aggregated data alone.
6. Conclusion
In summary, allo-HSCT should be considered when drug therapy is ineffective or intolerable. Genetic mutation profiles may serve as a decision-making tool. When an optimal HLA-matched donor is not available, an HLA-mismatched donor may be acceptable. Use of ATG or PTCy may enable safer allo-HSCT. Ruxolitinib may be effective for both controlling GVHD and managing VEXAS-related inflammatory symptoms. Further accumulation of cases is necessary to establish a treatment strategy for VEXAS syndrome.
Acknowledgments
This work was supported by the Japan Agency for Medical Research and Development (AMED) (23tk0124003h0001 to Yasuhito Nannya and Seishi Ogawa, 23ck0106875h0001 to Yasuhito Nannya) and the Japan Society for the Promotion of Science (JSPS); Scientific Research on Innovative Areas (24H00009 to Seishi Ogawa). The super-computing resource was provided by the Human Genome Center, the Institute of Medical Science, the University of Tokyo.
Author Contribution – per CRediT
Methodology: Kentaro Nagae (Equal), Hiroyuki Muranushi (Equal). Formal Analysis: Kentaro Nagae (Equal), Hiroyuki Muranushi (Equal). Investigation: Kentaro Nagae (Equal), Hiroyuki Muranushi (Equal). Writing – original draft: Kentaro Nagae (Lead). Resources: Kentaro Nagae (Equal), Hiroyuki Muranushi (Equal), Yasuhito Nannya (Equal). Conceptualization: Hiroyuki Muranushi (Lead). Writing – review & editing: Hiroyuki Muranushi (Equal), Yasuhito Nannya (Equal), Takeshi Maeda (Equal). Supervision: Takeshi Maeda (Lead).
Competing of Interest – COPE
The authors have no relevant financial or non-financial interests to disclose.
Ethical Conduct Approval – Helsinki – IACUC
This case report was created in line with the principles of the Declaration of Helsinki. Approval was granted by the Ethics Committee of Kurashiki Central Hospital (No. 4703).
Funding
No funding was received for conducting this study.
Consent to participate / Consent to publish
The participant has consented to the submission of the case report to the journal.